The purpose of this continuing professional development supplement is to explain how an integrated summary report can be created to consolidate the information required for assessment of immunogenicity-related risks of different types of biopharmaceuticals.

What is immunogenicity?
The term “immunogenicity” refers to the capacity of a therapeutic agent to induce an immune response in the recipient. The nature of the immune response depends on an interaction of multiple factors, including the structural features of the therapeutic agent, genotypic and phenotypic characteristics of the recipient and the particular conditions of use.[1],[2]
Although the administration of therapeutic proteins generally results in a detectable immune response – in the form of anti-drug antibodies (ADA) and drugspecific B/T-lymphocytes in the systemic circulation – this may often be without any impact on the overall clinical benefit versus risk balance. Nevertheless, in rare cases, treatment-emergent ADA has reacted with both the therapeutic agent and endogenous factors leading to severe, life-threatening consequences.[3]Aside from potential risk associated with a treatmentinduced immune response, human subjects may have pre-existing antibodies resulting from prior exposure to molecules that share common features with those of the therapeutic agent. Administration of the therapeutic agent against such a pre-sensitised background may then provoke serious adverse reactions in some subjects.[4]
Immunogenicity can be relevant for both sides of the benefit versus risk equation because ADA can reduce efficacy by enhancing clearance or by directly neutralising the activity of the therapeutic agent, as well as inducing immune-mediated adverse reactions.
Which types of medicinal product fall within the scope of this article?
This article concerns information relating to the assessment of undesirable immunogenicity of biopharmaceutical products to be submitted in support of clinical trial applications (CTA) and marketing authorisation applications (MAA) in International Council for Harmonisation (ICH) regions. The principles are applicable to originator and biosimilar versions of therapeutic proteins and peptides, as well as to gene and cell-based therapies. Vaccine products, for which immunogenicity is the intended therapeutic effect, are outside the scope of this article.
Why is this topic important for regulatory scientists?
There are a number of important activities for the regulatory specialist, most notably:
- Justification of the adequacy of the immunogenicity risk-based programme in regulatory submissions, including briefing material to support scientific advice meetings, CTA and MAA for most types of biopharmaceutical products
- Close collaboration with chemistry, manufacturing and controls (CMC) specialists to identify potential risks at the earliest stage of development and align manufacturing and product quality control decisions with an effective risk mitigation strategy
- Understanding of the suitability of bioanalytical methodology applied to monitor immune responses in nonclinical and clinical studies, including regulatory expectations for method validation
- Provision of advice to clinical team regarding sample timing for ADA testing and measurement of other relevant parameters (drug concentration, biomarkers of pharmacodynamic [PD] response, adverse events of special interest, etc) in clinical studies
- Incorporation in the statistical analysis plan of relevant terminology and data outputs for presentation in the MAA
- Integration of bioanalytical signals from nonclinical and clinical studies with relevant pharmacokinetics (PK), PD, efficacy and safety indices to describe the impact of immunogenicity on overall benefit versus risk
- Linkage of conclusions from clinical immunogenicity evaluation with other sections of the MAA dossier, including the risk management plan
- Updating of the immunogenicity summary to support authorisation in other therapeutic settings and maintenance of accuracy of prescribing information in respect of immunogenicity-related risks.
What is the ISI?
The ISI is intended to represent the “complete story” of how the applicant has evaluated and mitigated immunogenicity-related risks for the investigational medicinal product. The term “integrated” refers here to the linkage of bioanalytical signals (ADA) with clinical endpoints (PK, PD, efficacy and safety) within each clinical study, and to align with the potential risks including product quality-related factors, rather than aggregation of clinical data across all studies. Thus, the ISI concept is distinct from that of the integrated summary of efficacy (ISE) or integrated summary of safety (ISS). The value proposition for the sponsor is that the ISI can provide the regulatory assessor with the requisite information to understand the nature and extent of the risk at both the population and individual subject levels, thereby reducing uncertainty for clinical trial approval or marketing authorisation.
The ICH M4E R2 guideline[5] defines the summary information to be included in Modules 2.7.2.4 and 2.5.3 of the CTD format, with the associated bioanalytical method validation reports and standard operating procedures (SOPs) located in Module 5.3.1.4. Clinical study reports (CSRs) in Module 5.3 will also contain results from individual studies.
Guidance from both the European Medicines Agency (EMA)2 and the US FDA6 endorse the integrated summary concept and extend the scope of information defined in the ICH M4E R2 guideline to include:
- Risk assessment, ie, identification of potential risk factors and rationale for evaluation and mitigation
- Critical discussion of control of product qualityrelated factors, with a focus on product-related variants and process-derived impurities that may influence immunogenicity
- Clearer navigation through the development and validation of bioanalytical methodology in relation to assays applied to individual clinical studies (ie, to link the information contained in the reports submitted in Modules 5.3.1.4 and 5.3)
- More extensive descriptive analyses of the relationship between ADA signals and clinical parameters.
Section 10 of the EMA guideline2 provides a list of topics to be addressed in the ISI, while the FDA guidance[6] identifies the most important elements for the regulatory reviewer – in a manner that is fully consistent with the EMA guidance. The case study associated with this article explains how the different elements can be assembled to build an effective ISI. A more detailed model format is described in a separate article.[7]
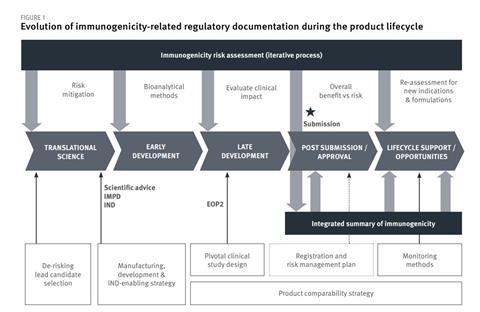
Where in the CTD format should the ISI be located?
Because the amount of information to be submitted may exceed size limitations for Module 2 summaries in the CTD format, both EMA and FDA guidelines2,6 explicitly permit submission of the ISI in Module 5.3.5.3. A brief summary of the results and conclusions of the clinical immunogenicity evaluation can be submitted in Module 2.7.2.4 and cross-refer to the ISI in Module 5.3.5.3 for the complete information.
In the author’s experience, the length of the ISI for a novel biological entity is often between 160 to 200 pages of A4 format. In the case of abbreviated programmes, eg, for biosimilars and line extensions, the ISI might be shorter and accommodated more easily in Module 2.7.2.4. Both the EMA and FDA have been flexible in allowing applicants to use their judgement about dossier location in order to encourage completeness of information to be reviewed.
How should the risk-based programme be described?
Regulatory guidance[2],[6] encourages the creation of a formal immunogenicity risk assessment document early in the product development process. A comprehensive framework of the scope of information to consider is provided in the EMA and FDA guidance[2],[6], bearing in mind that the risk assessment needs to be adapted to both the specific structural and functional properties of the investigational medicinal product (IMP) and the intended clinical population. The aim of the risk assessment is to identify potential product and patientrelated risk factors and thereby design appropriate monitoring and control measures into the development programme.
A summary report can be created to serve as a source document for information to be included in CTAs and briefing documents for scientific advice procedures. This document should be updated during clinical development and then used as the first section of the ISI for submission in the MAA dossier. Figure 1 illustrates the iterative process of immunogenicity risk assessment process as manufacturing process and clinical development progresses towards the MAA and linkage to the ISI.
Currently, there is no standard format for the risk assessment document. The format summarised in Figure 1 of the case study could be used as a starting point because this incorporates sub-headings that reflect the different points to consider described in the relevant regulatory guidelines.
[1],[2],[5],[6] The immunogenicity risk assessment may be submitted in the CTA dossier. In terms of the CTD format, section 2.7.2.4 would be the most relevant location for this information. Alternatively, it could be submitted as a sub-section of the overall benefit–risk justification for the clinical study, or as an appendix to the CTA dossier. It is recommended that sponsors seek endorsement from regulatory agencies about choice of methodology and clinical study design for monitoring immunogenicity. Even if validated bioanalytical methods are not yet available, the CTA dossier should provide a justification for the proposed extent of monitoring and bioanalytical methods to be developed and validated.
Which factors contribute most to successful regulatory outcomes?
Immunogenicity depends on the intrinsic molecular features of the IMP and the manner in which these interact with the innate and adaptive immune systems, against a background of variable immune tolerance.[8] Accordingly, explaining how molecular design and mode of action of the product (and its target) contribute to immunogenic potential in the relevant therapeutic context represents the foundation for the risk profile.
Because fundamental choices made at the levels of molecular design and host cell substrate to be used for expression of a recombinant protein can be critical variables, the rationale for and impact of the decisions made merits clear discussion in the ISI. Although not being accurately predictive of immune responses across a clinical population, in silico and in vitro tools are available that allow a first estimation of relative intrinsic immunogenic potential of a defined primary amino acid sequence[9].[10] [11]Therefore, presentation of data generated using these tools can provide a starting point for assessing relative risk.
Since formulation decisions can influence immunogenicity by affecting the stability of the monomeric form of the investigational medicinal product, and host cell-derived impurities can contribute to incremental immunogenicity, it is relevant to provide a summary of analytical and stability test results for such attributes in the ISI – or, at least, to provide a cross-reference to the relevant data located in Module 3 of the dossier.
The bioanalytical methods applied to monitor the ADA response generate an indirect index of immunogenicity, which is inferred from the difference between pre- and post-treatment signals in ligandbinding assays that measure antigenicity.
These assays are not quantitative because there are no absolute reference standards for calibration and the assays are subject to many other sources of analytical bias. Most importantly, these assays often detect signals at levels that are well below that associated with a clinical impact.[12]Therefore, the most important benefit of the ISI is the opportunity to position the bioanalytical signals into a relevant clinical context by integrating the results from ADA testing with indices of PK, PD, efficacy and safety. This requires advance planning to define the requisite data inputs for the ISI – which can be defined either in the statistical analysis plan for the clinical study (and, therefore, captured in the tables, figures and listings in the clinical study report) or in a pre-defined secondary data analysis plan (sometimes referred to as the “ISI SAP”) that is not part of the formal clinical protocol. Industry publications provide advice for terminology and data presentation that can be used for defining suitable analyses.[13],[14]
While inclusion of detailed information on bioanalytical methodology in the ISI might seem to duplicate content in the method validation reports to be submitted in Module 5.3.1.4, it can be extremely difficult for the regulatory assessor to follow the evolution of methodology applied to different clinical studies. Accordingly, the bioanalytical section of the ISI can be used to navigate the regulatory assessor through the history, clarifying how changes in methodology have affected assay performance/ADA detectability.
Given the important influence of genotypic and phenotypic variables on the magnitude of immune responsiveness, the results of the clinical immunogenicity evaluation should include a critical review of the impact at both the individual subject and population levels. In particular, comparison of clinical parameters (PK, PD, efficacy and safety) for the subpopulations with the lowest versus highest ADA titer values can be instructive for identification of subjects at highest risk. Appropriate warnings and precautions may then be included in the prescribing information.
Conclusion
The ISI represents a “living document” that provides a complete picture of the immunogenicity-related information that will need to be assessed by regulatory reviewers. This format can be created early in the development cycle to guide strategic decisions and then updated during clinical development to support regulatory interactions, initial marketing authorisation and line extensions for new therapeutic indications.
References:
[1] Guidance for industry: immunogenicity assessment for therapeutic protein products, FDA, August 2014. Available at https://www.fda.gov/ucm/groups/fdagov-public/@fdagovdrugs-gen/documents/document/ucm338856.pdf (accessed 10 December 2019).
[2] Guideline on immunogenicity assessment of therapeutic proteins (EMEA/CHMP/BMWP/14327/2006 Rev 1). Committee for Medicinal Products for Human Use (CHMP) May 2017. Available at http://www.ema.europa.eu/docs/en GB/document library/ Scientific guideline/2017/06/WC500228861.pdf (accessed 10 December 2019).
[3] Casadevall N, Eckhardt KU, Rossert J. Epoetin-induced autoimmune pure red cell aplasia. J Am Soc Nephrol 2005;16 Suppl 1:S67–9.
[4] Chung CH, Mirakhur B, Chan E et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med 2008;358(11):1109–17,
[5] ICH M4E R2 Common technical document for the registration of pharmaceuticals for human use – efficacy, Step 5, 15 July 2016. Available at https://www.ema.europa.eu/en/ich-m4e-commontechnical-document-registration-pharmaceuticals-human-useefficacy (accessed 10 December 2019).
[6] Guidance for industry: immunogenicity testing of therapeutic protein products – developing and validating assays for antidrug antibody detection, FDA January 2019. Available at https:// www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/ documents/document/ucm629728.pdf (accessed 10 December 2019).
[7] Chamberlain P. Effective presentation of immunogenicity risk assessments and related data in regulatory dossiers. Bioanalysis 2019 Feb 15. [Epub ahead of print]. Open Access article available at https://www.future-science.com/doi/10.4155/bio-2018-0209 (accessed 10 December 2019).
[8] Baker MP, Jones TD, Chamberlain P. Immunogenicity of biologics, In: Protein Therapeutics (eds Vaughan T, Osbourn J and Jallal B). Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2017.
[9] Jawa V, Cousens LP, Awwad M et al. T-cell dependent immunogenicity of protein therapeutics: preclinical assessment and mitigation. Clin Immunol 2013 Dec;149(3):534–55.
[10] .Joubert MK, Deshpande M, Yang J et al. Use of in vitro assays to assess immunogenicity risk of antibody-based biotherapeutics. PLoS One. 2016 Aug 5;11(8):e0159328.
[11] Gokemeijer J, Jawa V, Mitra-Kaushik S. How close are we to profiling immunogenicity risk using in silico algorithms and in vitro methods? An industry perspective. AAPS J 2017;19(6):1587– 1592.
[12] Song S, Yang L, Trepicchio WL et al. Understanding the supersensitive anti-drug antibody assay: unexpected high antidrug antibody incidence and its clinical relevance. J Immunol Res. 2016;2016:3072586.
[13] Shankar G, Arkin S, Cocea L et al. American Association of Pharmaceutical Scientists. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptidesharmonized terminology and tactical recommendations. AAPS J 2014;16(4):658–673.
[14] Rup B, Pallardy M, Sikkema D et al. Standardising terms, definitions and concepts for describing and interpreting unwanted immunogenicity of biopharmaceuticals: recommendations of the Innovative Medicines Initiative ABIRISK consortium. Clin Exp Immunol 2015;181(3):385–400.