






This continuing professional development article addresses changes introduced by Regulation (EU) No 536/2014 (the Clinical Trial Regulation/CTR) and focuses on the revised submission process for clinical trial applications in the EU/EEA via the new EU portal
Clinical trials performed in one or more EU/European Economic Area (EEA) countries are currently governed by Directive 2001/20 EC.[1] In accordance with this Directive, a clinical trial can only start when it has been authorised by the appropriate bodies (one or more competent authorities and one or more ethics committees). This Directive aimed to simplify and harmonise the administrative provisions governing clinical trials. Although there has been some alignment of clinical trial authorisations in the EU, experience has shown that a harmonised approach to the regulation of clinical trials has only been partly achieved. Regulation (EU) No 536/2014[2] intends to further increase a harmonised approach, thus creating an environment that is more favourable to conducting clinical trials in the EU.[3] The interpretation of the Regulation is explained in a draft Q&A document, which is being regularly updated.[4]

The aims of the Regulation are two-fold:
1. To achieve an internal market as regards clinical trials and medicinal products for human use (ie, to provide consistent rules for conducting clinical trials throughout the EU), taking as a base a high level of protection of health.
2. To set high standards of quality and safety for medicinal products in order to meet common safety concerns as regards these products.
To achieve this in the most efficient way and reduce administrative burden, the Regulation requires a single entry point for submitting clinical trial information in the EU/EEA (the portal; Article 80)2 and a database that contains all the data and information that is submitted and exchanged between parties (Article 81).[2] The system must allow regulatory oversight of clinical trials and provide collaboration tools, workflow and document management capabilities, accessible via individual workspaces.
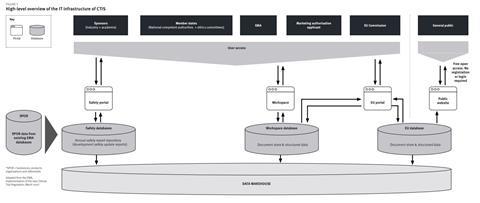
Once implemented, the Clinical Trial Regulation (CTR) will trigger major changes in existing processes for trial sponsors, both in the pharmaceutical industry and in academia. Changes will occur in the review and authorisation procedure (for both the initial application and substantial modifications), information on the trial that will be made publicly available and on safety reporting. However, the highest operational impact is expected from the introduction of the Clinical Trials Information System (CTIS). The CTIS will provide the single entry point for submitting, assessing, authorising, supervising and reporting a clinical trial in all member states in the EU/EEA. Figure 1 illustrates the high-level CTIS infrastructure.[5]

The CTIS will provide two secure workspaces (user interfaces); one for sponsors and one for member states. Separately, there will be an open access website, where clinical documentation and data are accessible to the public. Sponsors and member states each work in their own secure workspace and the EU portal is the “bridge” between these two secure workspaces that enables the transfer of documentation and data between them. The EU database will contain all information on trial applications on which a decision has been made by the member state. A separate safety reporting module will enable the submission and assessment of the annual safety report.
Underlying the databases is the data warehouse, which allows synchronisation of data in the databases and is used for reporting purposes. The CTIS is also connected to general databases that centralise management of four domains of master data, known collectively as SPOR[6] (see explanatory boxed text). On a practical level, the information contained in the data warehouse is used by member states’ inspectorates for managing inspections and by the European Commission for controlling purposes (oversight/monitoring for compliance of member states with the CTR).
Table 1 lists the key regulatory changes triggered by the CTR and it is immediately obvious that a component of the CTIS plays an important role in all but one of the key regulatory changes. A fully functional CTIS is deemed crucial to the success of the CTR, and hence as development of the CTIS is progressing, the go-live date is assumed to be December 2021 (status as of December 2020). The European Medicines Agency (EMA) is now publishing regular newsletters that can be subscribed to, with information and updates on the CTIS status.[7]
Implementation
The first step in the process is to ensure that all (potential) CTIS users are registered and have been assigned the appropriate profiles and roles they need for their work. A Sponsor Administrator will be assigned via the EMA identity and access management. Profiles for other users within the company will be assigned on a hierarchical basis within the sponsor organisation from the Sponsor Administrator role. Users that already have an EMA account only need to obtain the appropriate profile. Before accessing the CTIS, users will need to be trained. Depending on the role, users may be able to see different levels of information. The most common roles for daily work are dossier Viewer, dossier Preparer and dossier Submitter. The roles are rigidly defined but can be overlapped to create a flexible user profile. For example, it is possible to be given secure access as a Viewer of only Part I of the CTA or only the Q-IMPD (quality investigational medicinal product dossier) or only the CT results.
However, access can be given as a Viewer of multiple (or all) parts of the CTA. Similarly, for Preparers and Submitters access can be given for a specific area or for all activities. The Preparer of a specific CTA part automatically has Viewer access to the corresponding part (ie, the Preparer of Part II automatically has Viewer rights to Part II). The assigned sponsor has access to all documentation of the trial. Importantly, the system also allows a Sponsor Administrator to assign roles to individuals outside their own organisation (eg, to a contract research organisation contracted to perform the submission).
TABLE 1 Key regulatory changes when moving from the Clinical Trial Directive to the Clinical Trial Regulation for submission of a multinational trial in the EU/EEA
| Clinical Trial Directive 2001/20 – EudraCT(a) | Clinical Trial Regulation 536/2014 – CTIS portal and database(b) | Impact |
|---|---|---|
| Multiple submissions required for multinational trial – submission following individual member state process, no harmonised dossier content (e-submission of structured data/PDFs and/or paperbased submission). | Single submission electronically to all member states via CTIS portal. | Centralised EU-wide submission process. Further standardised submission documentation defined by the CTR. |
| Double submission within each member state: one to the regulatory authority (RA) and one (or more) to ethics committee(s) (EC). | ||
| Individual assessment by each member state without any IT collaboration tools available.(a) | Joint assessment of Part I scientific dossier by all member states together, facilitated by IT collaboration tools in the CTIS portal. Sponsor has the option to request the lead reviewer member state at time of application. | EU-wide member state collaboration. One harmonised assessment report and questions and comments on the Part I scientific dossier. Clearly defined timelines. |
| No single member state decision (RA and EC decisions separate, in some cases sequential). | One decision per member state (RA and EC) on Part II country-specific dossier components via the CTIS portal. | Harmonised country approval (EC and RA) on Part II country-specific dossier components at the same time, following defined timelines. |
| CTA fees charged per RA and EC. | One fee per member state review. | Reduced administration. |
| Limited information available to the public: structured data supplied via EudraCT application form and summary of results. | Subject to far-reaching transparency policies, possible to view and download CTA submission related content via the open access public website, eg, clinical study reports, full protocols; exceptions for commercially confidential information (CCI)/personal data (PD). | Heightened transparency requires sponsors to be aware of CTA contents that could be considered PD or CCI. |
(a) Does not apply to the voluntary harmonisation procedure (VHP)
(b) Appendix, on disclosure rules, to the “Functional specifications for the EU portal and EU database to be audited – EMA/42176/2014”[8]
Organisations (eg, trial sponsor details and all clinical trial sites) need to be registered through the OMS and this registration must be up to date. It is the responsibility of the sponsor to ensure clinical trial sites are registered or to register them.
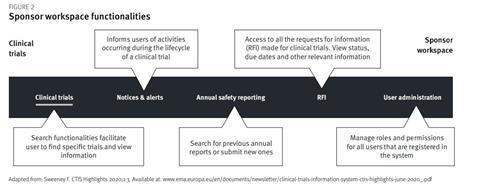
To submit the initial CTA, a user needs to access the sponsor workspace (Figure 2)[9]. The preparation of the CTA in the work area is started as draft and data can be collected and completed over time until the package is final and submission-ready. The functionalities can be accessed via the respective tabs. Under the tab “Clinical Trials” it will be possible to submit the initial application, submit documents to update the clinical trial dossier, add additional member states (only possible before or after an ongoing assessment) or submit notifications such as start of trial, start of recruitment, end of trial or summary of results. After initial authorisation, substantial and non-substantial modifications can be submitted in the same way. Under the tab “RFI”, users can see requests for information (RFI) from the member state, deadlines for responses and prepare/submit responses to the RFI. Under the tab “Annual Safety Reporting” a user can submit new annual safety reports or search for previously submitted ones. The tab “Clinical Trials” offers search functionalities that facilitate finding and viewing information on specific trials. Under “Notices & Alerts”, all activities that occurred during the lifecycle of a trial can be viewed, eg, under this tab it will be possible to view all decisions related to the clinical trial (for traceability or regulatory supervision with inspection reporting).
The application will be validated, assessed and decided upon in the member state workspace. Daily activities on assessment of a clinical trial application will be supported by the use of a workflow. Collaboration tools have been developed to facilitate communication among member states and with the sponsor representative.
Training
Users of the CTIS should be properly trained to acquire the necessary skills and knowledge to make optimal use of the system. The production of the first batch of online training modules started in early 2020 and is developing as planned.[10]The online approach will allow users to undertake the training at their own pace and convenience. In addition to the online training, the EMA intends to follow a train-the-trainer approach, whereby representatives of user groups will be trained as master trainers and will disseminate their knowledge within the group.[7]
Working in the transition period
The transition period lasts for three years. In the first 12 months, a CTA may still be submitted under the regulatory framework governed by Directive 2001/20/EC, or a CTA may be submitted in the CTIS and governed by the CTR. In the following 24 months, all initial CTAs must be submitted in the CTIS and are therefore governed by the CTR. Three years post go-live, all CTAs will be governed by the CTR, regardless of their date of submission, so any study that was started under the Directive and is still ongoing will have to be transitioned into the CTIS (ie, the sponsor has to collect the submission package of documents and upload it to the CTIS in order for the study to continue). Very old studies, ie, those that were submitted under national regulatory frameworks in place before the Directive was implemented, are unable to transition and may no longer continue under the CTR framework.
What are the next steps?
At present, the EMA CTIS project team is working on further improvement of functionalities related to the evaluation of a CTA, data submission and the public portal.7 A small number of nominated product owners (POs) representing member states and sponsors are contributing to the improved functionalities. The CTR requires that an independent audit is performed to verify that the CTIS has achieved full functionality and meets the functional specifications. The audit methodology was endorsed by the EMA’s Management Board in March 20207 and, subsequently, in June 2020 the EMA project team endorsed the audit scope. An external business consultancy has completed an independent audit of the EU portal and database (EUPD). If, on the basis of the audit report, the EMA Management Board and the European Commission agree that the conditions are met, the Commission will publish a notice in the Official EU Journal and the Regulation (EU) No 536/2014 will come into effect six months thereafter. As a working assumption the CTIS go-live date (and start of the transition period) is assumed to be December 2021.7 Further enhancement is still expected and planned after the go-live date, eg, the user experience features will be further strengthened.
Conclusion
The centralised EU-wide submission process (single submission of one CTA to all participating member states) that will be introduced with Clinical Trial Regulation 536/2014 is expected to have a high impact on daily clinical trial management processes. As initial CTIS implementation for member states and sponsors will be complex and challenging, thorough and timely preparation will be needed. Therefore, every future user should plan to invest sufficient time and effort to become familiar with the CTIS as it will become a part of their daily work. However, the anticipated benefits for all parties (such as extensive reporting possibilities, metrics overviews, harmonised submission requirements and assessments and extensive digitalisation of CTAs) are considerable and tangible – and are expected to repay the efforts invested.
[1] Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the member states relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Available at: https://ec.europa.eu/health/sites/health/ files/files/eudralex/vol-1/dir_2001_20/dir_2001_20_en.pdf (accessed 21 January 2021).
[2] Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/ EC. Available at: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2014_536/reg_2014_536_en.pdf (accessed 21 January 2021).
[3] EMA. Clinical Trial Regulation. Available at: https://www.ema. europa.eu/en/human-regulatory/research-development/clinicaltrials/clinical-trial-regulation (accessed 17 September 2020).
[4] The rules governing medicinal products in the European Union VOLUME 10 – Guidance documents applying to clinical trials. Clinical Trials Regulation (EU) No 536/2014 Draft Questions & Answers Version 2.6. Available at: https://ec.europa.eu/health/ sites/health/files/files/eudralex/vol-10/regulation5362014_qa_ en.pdf (accessed 21 January 2020).
[5] EMA. Implementation of the new Clinical Trials Regulation, March 2017. Available at: https://www.ema.europa.eu/en/documents/ presentation/presentation-key-features-objectives-regulation-euno-536/2014-laura-pioppo_en.pdf (accessed 26 January 2021).
[6] EMA. Data on Medicines (ISO IDMP Standards) - Substance, product, organisation and referential (SPOR) master data. Available at: https://www.ema.europa.eu/en/human-regulatory/ research-development/data-medicines-iso-idmp-standards/ substance-product-organisation-referential-spor-master-data (accessed 17 September 2020).
[7] EMA. CTIS highlights. Available at: https://www.ema.europa. eu/en/news-events/publications/newsletters#clinical-trialsinformation-system-(ctis)-highlights-section (accessed 21 January 2021).
[8] EMA/228383/2015 Endorsed. 2 October 2015. Appendix, on disclosure rules, to the “Functional specifications for the EU portal and EU database to be audited – EMA/42176/2014. Available at: https://www.ema.europa.eu/en/documents/other/ draft-appendices-draft-proposal-addendum-transparencyfunctional-specifications-european-union_en.pdf (accessed 21 January 2021).
[9] Sweeney F. CTIS Highlights. 2020;1:3. News, views and interviews for the Clinical Trials Information System (CTIS). Available at: https://www.ema.europa.eu/en/documents/newsletter/ clinical-trials-information-system-ctis-highlights-june-2020_.pdf (accessed 21 January 2021).
[10] CTIS Highlights. News, views and interviews for the Clinical Trials Information System (CTIS) 2020;2:4. Available at: https:// www.ema.europa.eu/en/documents/newsletter/clinical-trialsinformation-system-ctis-highlights-december-2020_en.pdf (accessed 21 January 2021).