Introduction
The purpose of this case study is to illustrate the creation of an integrated summary of immunogenicity report (ISI) that fulfils the recommendations described in current regulatory guidance for the scope of information needed to facilitate assessment of immunogenicity-related risks to support marketing authorisation of novel and biosimilar versions of biopharmaceutical products. A model format for documenting the output of the immunogenicity risk assessment that can be presented to support clinical trial applications and scientific advice procedures is also described. An updated version of this risk assessment may then used as the first part of the ISI to be submitted in the marketing authorisation application (MAA) dossier.

1. Immunogenicity risk assessment
The primary purpose of the risk assessment is to identify potential risks for the molecular entity and the target clinical population, and then to define an appropriate plan for evaluation and mitigation of these potential risks. Creation of a formal document that summarises the intrinsic and extrinsic factors that could influence the immunogenicity of an investigational medicinal product should commence at the time of the lead candidate selection stage of development because this will guide important risk evaluation and mitigation decisions. This document serves as the source for summaries to be presented in the briefing books in pre-CTA meetings and for the CTA dossier itself. The example in Figure 1 has been used by the author and could serve as a starting point for refinement and addresses the scope of information recommended in the regulatory guidance referred to in the main article.
For biosimilar applications, the analysis of risk factors would be guided primarily by the experience accumulated for the originator product allied to potential for incremental risk associated with any detected qualitative and quantitative differences in the product quality profile of the biosimilar candidate. Representative analytical results pertaining to potentially important variables for incremental immunogenicity risk may also be included. Nonclinical evaluation includes data from in vitro analyses using human cells and, for products that have structural and functional commonality across different species, in vivo data from non-human species might be relevant for hazard identification. The forward-looking plan for risk evaluation should include clinical study design considerations, as well as proposals for bioanalytical methodology.
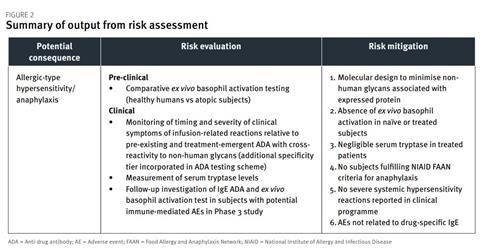
Figure 2 illustrates the way in which a tabular summary format could be used to present the output of the risk assessment process for one potential consequence of immunogenicity for a therapeutic protein expressed in a yeast host cell substrate. The risk identification process links to the definition of a suitable bioanalytical strategy for monitoring pertinent risks, as well as clinical trial design considerations, which can then be discussed with regulatory agencies as part of scientific advice procedures during development.
2. Integrated summary of immunogenicity (ISI)
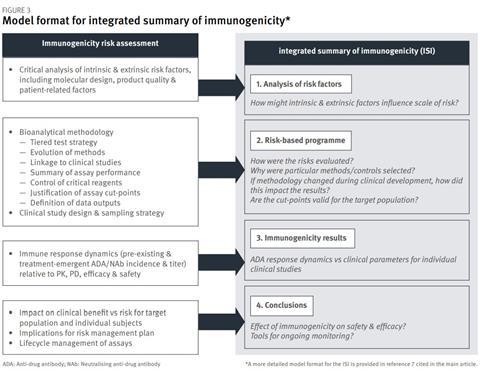
Regulatory guidance defines the scope of information to be presented in the ISI in a manner that allows flexibility to accommodate different product types, risk profiles and extent of the programme. The outline format shown in Figure 3 identifies the recommended inputs, including the updated risk assessment (Section 1 of ISI), and illustrates how these can be organised to address regulatory expectations.
Section 2 of the ISI provides an opportunity to explain the suitability of the methodology applied for risk evaluation at different stages of clinical development – what methods were chosen, and why – and how the data were interpreted. Effectively, Section 2 of the ISI can draw together the most critical information from the bioanalytical method validation reports submitted in Module 5.3.1.4, and help the regulatory assessor understand how different iterations of methodology fit into the clinical development programme.
Section 3 of the ISI should present the results from each clinical study in a manner that describes immune response dynamics (incidence and magnitude of anti-drug antibody/neutralising anti-drug antibody) relative to clinical points (pharmacokinetics, pharmacodynamics, efficacy and safety). Section 4 of the ISI provides the main conclusions and links to the risk management plan.
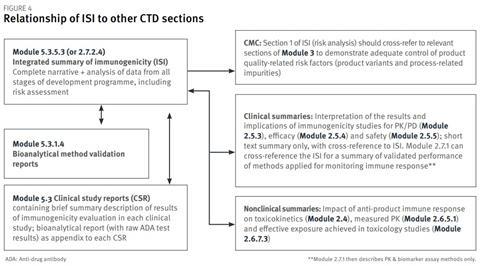
Figure 4 indicates the relationship of the ISI to other sections of the CTD format that are relevant for the immunogenicity assessment. Summary information and conclusions from the ISI can be used to populate clinical summaries. ISI section 1 will cross-refer to relevant sections of Module 3 for product qualityrelated information; ISI section 2 will cross-refer to the bioanalytical method validation reports in Module 5.3.1.4; and ISI section 3 will cross-refer to the clinical study reports in Module 5.3.