









Regulatory Rapporteur
February 2026 | Volume 23 | No. 2
Abstract
The In Vitro Diagnostic Regulation 2017/746 (IVDR) reshaped the regulatory landscape for in vitro diagnostic manufacturers and clinical trial sponsors conducting clinical trials across the European Union. By introducing stricter requirements for clinical evidence, performance evaluation and risk classification, the Regulation raised the bar for product safety and scientific rigour. Sponsors face increasing regulatory complexity, particularly when performance studies are conducted alongside clinical trials of investigational medicinal products under EU Clinical Trial Regulation 536/2014 (CTR).
This article examines common regulatory feedback and documentation requests arising from clinical performance study submissions, drawing on recent requests for information (RFIs) from both competent authorities and ethics committees. The authors, subject matter experts in the EU CTR/EU IVDR interface from IQVIA’s Clinical Trials Regulatory Management (CTRM) group, draw on internal regulatory intelligence to analyse recurring themes, such as risk analysis, document consistency, scientific validity and language clarity, and offer practical guidance to help sponsors and regulatory professionals navigate dual submissions under IVDR and CTR. Actionable recommendations and summaries are provided for each key document type to support compliance, reduce delays and improve submission quality.
Licence notice
Copyright © 2015-2026 The Organisation for Professionals in Regulatory Affairs Ltd. T/A Regulatory Rapporteur − All Rights Reserved. This work is licensed to Amber McNair and Vladimir Vujovic of IQVIA for commercial use.
Notwithstanding this licence, no part of materials published in Regulatory Rapporteur may be reproduced without the express written permission of the publisher.
As a general rule, permission should be sought from the rights holder to reproduce any substantial part of a copyrighted work. This includes any text, illustrations, charts, tables, photographs, or other material from previously published sources.
To obtain permission(s) to re-use content published in Regulatory Rapporteur please email publications@topra.org.
To join TOPRA please click here.

FOCUS: Navigating regulatory scrutiny: An analysis of requests for information in EU combined trials
DOWNLOAD
Introduction
The introduction of Regulation (EU) 2017/746 for in vitro diagnostic (IVD) medical devices has significantly transformed the regulatory environment for IVD manufacturers and clinical trial sponsors throughout the EU. With a focus on strong clinical evidence, risk-based classification and thorough performance evaluation, IVDR aims to enhance patient safety and public health protections.
However, transitioning to this new framework has posed substantial regulatory challenges, especially concerning clinical performance studies (CPS) in an investigational medicinal product (IMP) clinical trial (known as ‘combined clinical trials’). Evolving guidance from the EU’s Medical Device Coordination Group (MDCG) and varying expectations across Member States have created a fragmented and unpredictable environment for sponsors conducting combined clinical trials. In response, the European Commission initiated the COMBINE Project,[1] an effort to harmonise regulatory processes for trials involving both IMPs and IVDs. By integrating the CTR and IVDR submission pathways within the Clinical Trials Information System (CTIS), the project aims to minimise duplication, streamline workflows and provide clearer guidance for sponsors navigating these complex studies.
Until the COMBINE Project’s pilot phase outcomes are fully evaluated, sponsors must continue to manage two separate submission processes, each with its own objectives, timelines and regulatory expectations, often placing strain on resources and study approval planning. A crucial aspect of the regulatory process for combined studies requiring dual submissions is the management of requests for information (RFIs).
Based on an extensive review of combined clinical trial submissions managed by IQVIA, and analysis of RFIs received from multiple EU Member States, this article explores RFIs issued by national competent authorities (NCAs) and ethics committees (ECs) during the review of performance studies under IVDR when in combination with a clinical trial submitted under CTR, and identifies recurring themes, regulatory challenges and areas of ambiguity. Drawing on real-world data and stakeholder experiences, it offers practical insights and strategies for proactive regulatory oversight. The analysis is grounded in an anonymised, categorised internal repository of RFIs and correspondence compiled by IQVIA’s CTRM group during validation and assessment phases of combined EU CTR/IVDR submissions. Findings are reported at aggregate level and are intended to highlight common issues encountered in combined study applications from the perspective of performance studies without reference to identifiable sponsors or trials.
This article aims to help regulatory professionals navigate the complexities of IVDR implementation in clinical trials and contribute to broader efforts to harmonise and streamline combined clinical trial processes across the EU.
The complexity of combined studies under CTR and IVDR
Combined studies involving both IMPs and IVDs present a unique set of regulatory challenges. Their complexity stems from the need to navigate multiple distinct regulatory frameworks. Each framework has its own authorisation pathways, documentation standards and timelines, which require sponsors to manage parallel processes that are not fully harmonised.
National variability further compounds this challenge. While CTR benefits from a coordinated assessment procedure across Member States, CPSs under IVDR are authorised nationally. For example, Germany often requires additional device-specific risk assessments and investigator training in accordance with local legislation, and Italy and Spain mandate translations of technical documentation for review, even if the CTR submission is in English. Austria often requires additional justification for the clinical relevance of the biomarker and may request separate insurance coverage for the IVD component. In Poland, for delegation of authority, wet-ink originals and sworn Polish translations are still required for core authorisations, nudging sponsors back towards paper-based workflows.
Procedurally, certain countries, such as Italy, Spain and Germany, mandate that submissions occur sequentially and the EC must review and approve the application before it is reviewed by the NCA. In contrast, many countries permit the EC and NCA reviews in parallel, allowing for simultaneous submissions which potentially expedites the overall approval process.
Additional procedural and content-related challenges have also been highlighted in the European Commission’s analysis report on combined CTR–IVDR–MDR studies.[2] This results in a significant procedural burden; sponsors must submit multiple applications for a single study, which increases administrative workload and delays study initiation. Moreover, combined studies must meet both scientific and ethical standards, which can differ substantially between IMPs and IVDs. Clinical trials focus on therapeutic efficacy and safety endpoints, where performance studies emphasise analytical and clinical performance metrics (for example, sensitivity and specificity), adding another layer of complexity to the review process.
NCAs and ECs routinely issue RFIs and, in combined submissions, where sponsors present documentation for both IMPs and IVDs simultaneously, our analysis of these requests finds inconsistencies, gaps or ambiguities in submission dossiers. These RFIs offer valuable insights into the practical challenges of implementing harmonised reviews and underscore the urgent need for clearer guidance and streamlined processes.
Most frequently queried documents in combined study RFIs
This section presents a structured analysis of the most frequently queried documents by NCAs and ECs during the validation and assessment of all CPSs when in a combined clinical trial design under IQVIA management since IVDR implementation in 2022.
Using a comprehensive review of RFIs received from multiple EU Member States, we identified which documents faced the most scrutiny. These are summarised in Table 1, showing the most common RFIs received in red, and moving down the scale to the least number of RFIs received.
Table 1: Tabular summary of the most frequent IVDR RFIs in combined clinical trials
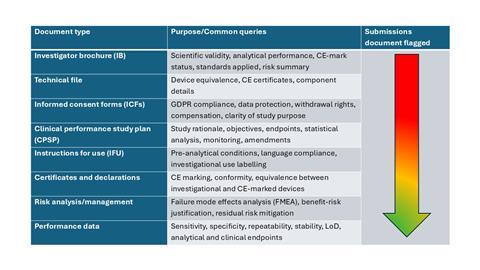
Investigator’s brochure (IB)
Authorities often request detailed risk-benefit analysis in the IB. Scientific validity and analytical performance data must be clearly documented, issuing comments such as:
‘Provide a demonstration of scientific validity and analytical and clinical performance as mentioned in Annex XIII Part A of EU Regulation 2017/746 in the Investigator’s Brochure; integrate the state of the art in the Investigator’s Brochure. Provide the list of standards applied (standards/standards applied in full, or in part, with justification on the parts not applied); This list must be integrated in the Investigator’s Brochure.’
Action points:
- Present comprehensive analytical validation summaries and peer-reviewed evidence supporting biomarker use
- Align with IVDR Annex XIII requirements
Clinical performance study plan (CPSP)
CPSPs must include standalone endpoints specific to the IVD. These endpoints must be distinct from those outlined in the clinical trial protocol and should directly relate to the analytical or clinical performance of the IVD.
Assessors commonly make the following comments:
‘The objective of the clinical trial being comparing efficacy of X is not compatible with what is expected to be the objective of a performance study. A performance study to evaluate the performance of IVD should be designed to evaluate clinical predictive performance of the test.’
‘Endpoints in the CPSP should be clearly defined and independent from those in the medicinal product protocol.’
Action points:
- Define clear analytical or clinical endpoints within the CPSP that reflect the performance objectives of the IVD
- Ensure these endpoints are documented independently of the drug protocol, particularly in combination studies, and are traceable to the scientific validity and risk analysis presented in the Investigator’s brochure
CE marking and conformity
Authorities often flag unclear CE status and intended use of IVDs, especially when used outside their certified scope. Feedback commonly received in these scenarios is as follows:
‘It would appear that IVDMD is CE-marked and used off-label. Please correct this section and submit all technical documentation, including the EU declaration of conformity and a valid CE marking certificate; Where applicable, provide information concerning the notified body.’
Action points:
- List all IVDs, their CE-marking status, intended use, and whether they are investigational
- Add this to the cover letter and protocol
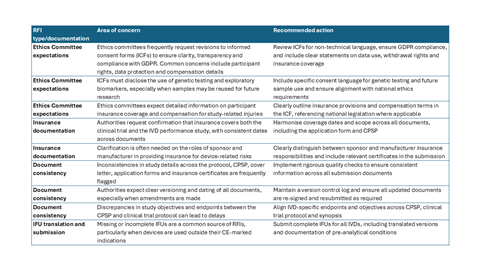
In Table 2, more examples of common RFIs received are provided.
Table 2: Summary of common regulatory and ethics committee RFIs in combined clinical trials
Managing RFIs under IVDR in the context of CTR submissions
Managing RFIs under IVDR presents distinct operational and strategic challenges.
A major complexity stems from the asynchronous review processes. The CTR benefits from a harmonised and centralised assessment procedure through CTIS, allowing coordinated feedback from Member States. In contrast, IVDR currently lacks a fully operational centralised infrastructure, like EUDAMED,[3] for performance study submissions. Consequently, CPSs are reviewed nationally, often with differing timelines, formats and expectations. This divergence means that RFIs related to the IVD component may be issued separately from the CTR review, creating fragmented feedback loops and complicating the sponsor’s ability to respond effectively.
Additionally, RFIs under IVDR are typically more technical, focusing on analytical validation, device-specific endpoints and performance characteristics. Coordinating responses across these teams while ensuring alignment with the CTR submission requires robust internal processes and clear ownership.
Sponsors also face complexity from document interdependence. For instance, a change to the CPSP in response to an IVDR-related RFI may require corresponding updates to the clinical trial protocol, informed consent forms, and application dossiers submitted via CTIS. Without a harmonised mechanism to manage these changes across both regulatory pathways, sponsors risk inconsistencies that can lead to further RFIs or delays.
To address these challenges, sponsors are increasingly adopting integrated RFI management strategies, including the following:
- Centralised tracking systems to monitor RFIs across jurisdictions and regulatory frameworks: For example, some sponsors have successfully implemented regulatory dashboards that track RFI status, deadlines and responsible teams in real time, thereby reducing missed timelines and improving accountability
- Cross-functional response teams to ensure timely and coordinated input: Effective models include forming regulatory touchpoints during critical review phases, where clinical, regulatory, quality and data management experts collaborate on responses. IQVIA case studies show that this approach can cut RFI turnaround times, especially when combined with predefined escalation protocols
- Documentation of harmonisation protocols to ensure consistency across all study materials: Assessment of substantiality and version control processes are key to the management of the harmonisation of documents post-multinational submissions under IVDR. For example, if Spain requested an update to the CPSP, but it has already been submitted and approval obtained by the EC in Italy, it may be possible to submit a harmonised CPSP at the end of the submission process to all Member States as a non-substantial amendment (rather than submitting an amendment to the Italian EC before proceeding with the NCA application)
Other successful methods of management of combined RFIs include the following:
- Pre-submission package reviews using regulatory experts to identify gaps likely to generate RFIs
- Training programmes for study teams on IVDR-specific requirements, ensuring proactive compliance rather than reactive fixes
As the European Commission considers harmonised submission planning under the EU Biotech Act, there is hope that future submissions will benefit from better alignment and reduced administrative burden. Until then, sponsors must navigate a complex and evolving landscape where effective RFI management is vital to maintaining study timelines and regulatory compliance.
Discussion: Navigating the regulatory complexity of combined studies
The examination of RFIs issued during the evaluation of combined clinical trials and performance studies under CTR and IVDR reveals a persistent and complex regulatory challenge. Although the goal of submitting a unified study package is to harmonise scientific and ethical evaluations,[4] the reality is often more fragmented. The absence of coordinated assessment procedures for CPSs, along with national differences in interpretation and implementation of IVDR, creates a situation where sponsors must anticipate and address varying expectations.
The most frequently questioned segments highlight a fundamental tension between the harmonised goals of EU legislation and the practical realities of its implementation. For example, NCAs and ECs frequently raise RFIs when CPSPs lack IVD-specific endpoints. This is not a minor detail; it is fundamental to demonstrating compliance with the general safety and performance requirements (GSPRs) under IVDR. Even in drug-diagnostic combination studies, regulators expect the IVD component to be independently justified and evaluated. This reflects both regulatory rigour and scientific logic: combined studies pursue distinct objectives. The medicinal product is assessed for therapeutic efficacy and safety, while the IVD is evaluated for diagnostic accuracy and reliability.
Endpoints for the drug typically include clinical outcomes, such as progression-free survival or response rates. For the IVD, endpoints must capture analytical and clinical performance metrics that confirm the device’s ability to accurately identify the target biomarker or patient population. Blending these endpoints risks conflating therapeutic effect with diagnostic utility, potentially compromising clarity and validity of the data.
Lack of harmonised templates or procedural standards for CPSPs exacerbates this issue, leaving sponsors to interpret and adapt requirements on a case-by-case basis. The emphasis on analytical validation, safety reporting and data retention[5] further reflects IVDR’s rigorous standards, which, while essential for ensuring device quality and patient safety, demand a level of documentation and procedural clarity that may exceed what is traditionally expected in medicinal product trials. The cumulative effect is a significant regulatory burden, with sponsors often required to submit multiple versions of documents tailored to individual Member State requirements. Perhaps the most telling is the frequency of RFIs related to inconsistencies across submission documents. These discrepancies, often minor, can trigger substantial delays and signal a broader need for integrated document management strategies and cross-functional collaboration during study preparation.
Considering these findings, it is evident that sponsors must adopt a proactive and strategic approach to combined study submissions. This includes rigorous internal harmonisation of documentation and the development of CPSPs that clearly articulate the IVD’s role, performance objectives and validation strategy. Ultimately, while combined studies offer significant scientific and operational efficiencies, their successful execution under the current regulatory framework requires careful planning, robust documentation, and ongoing dialogue between sponsors, regulators and ethics committees. As the EU continues to refine its regulatory infrastructure,[6][7] there is an opportunity to address these challenges and move towards a more integrated and predictable review process for combined studies.
Conclusion: Toward greater clarity and coordination in combined study submissions
Sponsors conducting combined studies under the CTR and IVDR frameworks provide valuable opportunities to advance precision medicine through integrated drug-diagnostic development. However, the analysis of RFIs highlights the practical challenges sponsors encounter when navigating two distinct regulatory pathways, each with its own scientific goals, documentation requirements and review processes.
A key takeaway from this analysis is the importance of maintaining separate and scientifically justified endpoints for the medicinal product and the in vitro diagnostic device. Although these components can be studied simultaneously, their regulatory and scientific objectives differ significantly. The medicinal product is evaluated for therapeutic effectiveness and safety, while the IVD must demonstrate diagnostic accuracy and performance. Sponsors need to clearly define these endpoints to ensure that each component is assessed on its own merits, supports regulatory compliance and strengthens the overall study.
The current lack of standardised templates and coordinated assessment procedures for clinical performance studies can lead to varying expectations across Member States. This increases the administrative complexity of submissions and requires additional effort to manage RFIs and maintain consistency in documentation. These processes uphold safety standards, but to maintain high standards of safety and performance, they can pose operational challenges that might affect study timelines and resource planning.
As the EU continues to develop its regulatory infrastructure, particularly with the anticipated EU Biotech Act, full functionality of EUDAMED and the expected outcomes of the EC COMBINE pilot, there is a promising opportunity to enhance coordination and predictability in the review of complex studies.[8] By promoting consistency in documentation and review standards, the EU Biotech Act represents a crucial step toward enabling innovation while ensuring patient safety and scientific integrity in complex trial designs. However, ongoing collaboration between sponsors, regulators and ethics committees will be essential to fully realise the potential of combined studies, supporting innovation while maintaining the integrity and safety of clinical research.
References
[1] European Commission ‘The COMBINE programme: Legislative context for COMBINE’. (Accessed: January 2026).
[2] European Commission (2024) ‘COMBINE CTR–IVDR–MDR: Analysis Phase Report’. doi:10.2875/138207. (Accessed: January 2026).
[3] European Commission (2024) ‘Gradual roll-out of EUDAMED – Q&As on practical aspects related to the implementation of Regulation (EU) 2024/1860’. (Accessed: January 2026).
[4] European Commission (2025) ‘MDCG 2025-5 - Questions & Answers regarding performance studies of in vitro diagnostic medical devices under Regulation (EU) 2017/746 (June 2025)’. (Accessed: January 2026).
[5] MedEnvoy (2025) ‘MDCG 2025-5: New IVDR Guidance on IVD Performance Studies’. (Accessed: January 2026).
[6] LexisNexis Reed Tech (2024) ‘EUDAMED Questions Answered; Navigating the Gradual Roll-Out of EUDAMED under MDR and IVDR Updates’. (Accessed: January 2026).
[7] MedQair (2024) ‘EUDAMED: Current Status, Timelines, and Implementation Guide’. (Accessed: January 2026).
[8] TOPRA (2025) ‘HM5/MD1/IVD1: Programme COMBINE – Accelerating clinical trials and medical innovation in the EU’. Regulatory Rapporteur. Vol. 22(10). (Accessed: January 2026).
With thanks to Christopher Bamford (IQVIA), Elizabeth Moir (IQVIA) and Sarah Johns (MCRA, an IQVIA business).
Access further Regulatory Rapporteur resources below:
-
HM5/MD1/IVD1: Programme COMBINE – Accelerating clinical trials and medical innovation in the EU (TOPRA Symposium 2025)
-
HM5/MD1/IVD1: COMBINE project: A new era of combined clinical trials in Europe (TOPRA Symposium 2024)
This article is featured in Regulatory Rapporteur Volume 23, No. 2